How to Create a Protein Dataset
A step-by-step guide to generating ESM3 embeddings for protein sequences, featurizing side chains, and organizing these features into a PyTorch Geometric dataset.
Introduction
If you’d like to follow along or run this notebook yourself, you can find both the notebook and dataset in my GitHub repository.
In this tutorial, we’ll learn how to generate ESM3 embeddings for a protein sequence, featurize its side chains, and store these features in a PyTorch Geometric dataset. If you’re not familiar with torch_geometric, you may find it helpful to review the previous tutorial How to Create a Molecular Dataset.
For this walkthrough, I prepared a dataset of six proteins, each with 3D structures in PDB format. Let’s start by importing the required libraries, downloading the dataset, and visualizing one of the protein structures (specifically, the one with PDB ID: 5HMK).
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
from Bio.PDB import PDBParser, is_aa
from Bio.SeqUtils import seq1
import py3Dmol
from esm.models.esm3 import ESM3
from esm.sdk.api import ESMProtein, SamplingConfig
from esm.utils.constants.models import ESM3_OPEN_SMALL
from torch_geometric.data import InMemoryDataset, Data
from torch_geometric.data import download_url, extract_zip
from torch_geometric.utils import one_hot
from huggingface_hub import login
import numpy as np
import torch
import os
1
2
3
! mkdir -p data
! wget -q https://github.com/vladislach/protein-dataset/raw/main/proteins.zip
! unzip -q -o proteins.zip -d data/raw; rm proteins.zip
1
2
3
4
5
6
7
8
9
10
11
def visualize_protein(pdb_path):
with open(pdb_path, 'r') as f:
pdb_data = f.read()
view = py3Dmol.view(width=500, height=350)
view.addModel(pdb_data, 'pdb')
view.setStyle({'cartoon': {'color': 'spectrum'}})
view.zoomTo()
view.show()
visualize_protein('data/raw/5hmk.pdb')
The structure above is interactive, so you can zoom in/out and rotate it.
ESM3 Embeddings
First, we’ll obtain ESM3 embeddings for the protein sequences. The latest ESM3 model includes a convenient Python API for generating these embeddings. You can find the model weights on the Hugging Face Hub at HuggingFace/EvolutionaryScale/esm3.
To use this API, you’ll need to have the esm and huggingface_hub installed (pip install esm huggingface_hub). Next, you’ll nedd to accept the ESM3 dataset’s terms and conditions on your Hugging Face account to enable the download of model weights (more details are available in Quickstart for ESM3-open). After accepting the license, run the code below to download the model weights and generate embeddings. Don’t forget to replace <TOKEN> with your Hugging Face API token for correct authorization.
Running this step on a machine with a GPU is recommended, as the model is large and may take some time to download and process.
1
2
login(token='<TOKEN>')
client = ESM3.from_pretrained(ESM3_OPEN_SMALL)
Now we can obtain the embeddings. The function below takes as input a path to the directory with all .pdb files and returns a dictionary mapping each protein name (PDB ID) to its ESM3 embeddings.
A couple things to note:
- We use
biopythonto parse the.pdbfiles and extract protein sequences. - In this tutorial, the
.pdbfiles are pre-cleaned and contain only protein residues. Typically, this may not be the case, so I included a commented-out check to remove any non-protein residues (line 17). You might also consider adding checks for non-standard amino acids if your dataset includes them. residue.get_resname()returns the three-letter amino acid code, which we convert to a one-letter code using theBio.SeqUtils.seq1function.- Some proteins contain multiple chains. In such cases, we treat each chain as a separate sequence, generate embeddings for each, and then concatenate them to get the final embeddings for the protein.
- The output embeddings have a shape of
[L + 2, 1536], whereLis the length of the protein sequence. The two additional embeddings are for the start (<cls>) and end (<eos>) sequence tokens in the model. We remove these two embeddings before storing the final result (line 26).
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
def get_esm_embs(data_dir: str) -> dict:
"""Get ESM3 embeddings for proteins in the given directory."""
names = [f.split('.')[0] for f in os.listdir(data_dir) if f.endswith('.pdb')]
paths = [os.path.join(data_dir, f) for f in os.listdir(data_dir) if f.endswith('.pdb')]
parser = PDBParser()
esm_embs = {}
for name, path in zip(names, paths):
structure = parser.get_structure(name, path)
chain_embs_list = []
for chain in structure.get_chains():
seq = ''
for residue in chain:
# if not is_aa(residue): continue
seq += seq1(residue.resname)
protein = ESMProtein(sequence=seq)
protein_tensor = client.encode(protein)
output = client.forward_and_sample(
protein_tensor, SamplingConfig(return_per_residue_embeddings=True)
)
chain_embs_list.append(output.per_residue_embedding[1:-1, :].cpu())
emb = torch.cat(chain_embs_list, dim=0)
esm_embs[name] = emb
return esm_embs
Let’s generate embeddings for our proteins and take a look at the embeddings for a sample protein in the dataset.
1
2
3
4
esm_embs = get_esm_embs('data/raw')
print(esm_embs['5hmk'].shape)
print(esm_embs['5hmk'][:5, :])
torch.Size([86, 1536])
tensor([[ 184.0000, 210.0000, 58.5000, ..., -111.0000, -81.0000,
-92.5000],
[ -94.0000, -116.0000, 138.0000, ..., -65.0000, -12.5000,
-69.5000],
[ 157.0000, 142.0000, 5.0625, ..., -80.0000, 62.7500,
-10.0000],
[ 146.0000, -28.5000, -136.0000, ..., -226.0000, 37.0000,
117.0000],
[ 66.0000, 65.5000, 26.7500, ..., 27.5000, 199.0000,
53.5000]])
We’ll save the embeddings in the data directory for future use:
1
torch.save(esm_embs, 'data/esm_embs.pt')
Sidechain Features: Chi Angles and Relative Positions
While the ESM3 embeddings capture global features of the protein sequence and can suffice for some tasks, we can also extract side chain features to capture local details of the protein. In this tutorial, we’ll extract the chi angles and relative positions of specific atoms in the side chains of the protein residues.
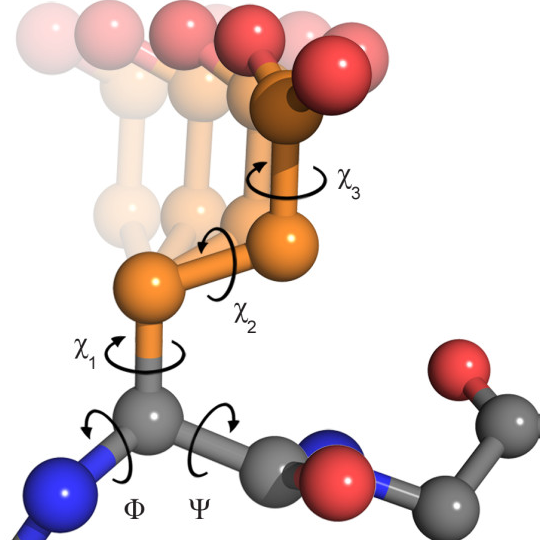
Figure 1. Illustration of chi angles (χ1, χ2, χ3) in a protein side chains.
The chi angles are dihedral angles of the side chain atoms and are important for understanding side chain conformation. Some amino acids (i.e., glycine and alanine) have no chi anlges because their side chains are too short to form a dihedral angle, while others have multiple chi angles (e.g., methionine). The table below summarizes the amino acids and the number of chi angles they have:
| Amino Acid | Rotamer Angles |
|---|---|
| Arginine (Arg, R) | χ1, χ2, χ3, χ4 |
| Lysine (Lys, K) | χ1, χ2, χ3, χ4 |
| Glutamine (Gln, Q) | χ1, χ2, χ3 |
| Glutamate (Glu, E) | χ1, χ2, χ3 |
| Histidine (His, H) | χ1, χ2 |
| Isoleucine (Ile, I) | χ1, χ2 |
| Leucine (Leu, L) | χ1, χ2 |
| Methionine (Met, M) | χ1, χ2, χ3 |
| Phenylalanine (Phe, F) | χ1, χ2 |
| Tyrosine (Tyr, Y) | χ1, χ2 |
| Tryptophan (Trp, W) | χ1, χ2 |
| Threonine (Thr, T) | χ1 |
| Serine (Ser, S) | χ1 |
| Valine (Val, V) | χ1 |
| Asparagine (Asn, N) | χ1, χ2 |
| Aspartate (Asp, D) | χ1, χ2 |
| Cysteine (Cys, C) | χ1 |
| Proline (Pro, P) | χ1 |
| Glycine (Gly, G) | - |
| Alanine (Ala, A) | - |
Now let’s create some useful mappings to help us extract the chi angles from the .pdb files. The first dictionary (atom_order) will convert the one-letter amino acid code to the atom order in the .pdb file (excluding hydrogens) for that amino acid. The atom names and their order are standardized in .pdb files for each amino acid, so this mapping will allow us to store the coordinates of individual atoms in the right order. The maximum number of atoms for any amino acid is 14.
[Some of these mappings are adapted from DiffDock/datasets/constants.py].
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
amino_acids = 'ACDEFGHIKLMNPQRSTVWY' # 20 standard amino acids, will be used for one-hot encodings later
atom_order = {
'G': ['N', 'CA', 'C', 'O'],
'A': ['N', 'CA', 'C', 'O', 'CB'],
'S': ['N', 'CA', 'C', 'O', 'CB', 'OG'],
'C': ['N', 'CA', 'C', 'O', 'CB', 'SG'],
'T': ['N', 'CA', 'C', 'O', 'CB', 'OG1', 'CG2'],
'P': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD'],
'V': ['N', 'CA', 'C', 'O', 'CB', 'CG1', 'CG2'],
'M': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'SD', 'CE'],
'N': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'OD1', 'ND2'],
'I': ['N', 'CA', 'C', 'O', 'CB', 'CG1', 'CG2', 'CD1'],
'L': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD1', 'CD2'],
'D': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'OD1', 'OD2'],
'E': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD', 'OE1', 'OE2'],
'K': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD', 'CE', 'NZ'],
'Q': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD', 'OE1', 'NE2'],
'H': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'ND1', 'CD2', 'CE1', 'NE2'],
'F': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD1', 'CD2', 'CE1', 'CE2', 'CZ'],
'R': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD', 'NE', 'CZ', 'NH1', 'NH2'],
'Y': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD1', 'CD2', 'CE1', 'CE2', 'CZ', 'OH'],
'W': ['N', 'CA', 'C', 'O', 'CB', 'CG', 'CD1', 'CD2', 'CE2', 'CE3', 'NE1', 'CZ2', 'CZ3', 'CH2']
}
The next mapping will associate each amino acid with its chi angles and the corresponding atoms that form these angles. For example, the \(\chi_2\) angle of arginine (R) is formed by the atoms 'CA-CB-CG-CD'. We will use this mapping to extract the correct coordinates to calculate the chi angles. Note that both glycine and alanine are excluded from this mapping because they have no chi angles.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
chi_atoms = {
'C': {
1: ('N', 'CA', 'CB', 'SG'),
},
'D': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'OD1'),
},
'E': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'CD'),
3: ('CB', 'CG', 'CD', 'OE1'),
},
'F': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'CD1'),
},
'H': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'ND1'),
},
'I': {
1: ('N', 'CA', 'CB', 'CG1'),
2: ('CA', 'CB', 'CG1', 'CD1'),
},
'K': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'CD'),
3: ('CB', 'CG', 'CD', 'CE'),
4: ('CG', 'CD', 'CE', 'NZ'),
},
'L': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'CD1'),
},
'M': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'SD'),
3: ('CB', 'CG', 'SD', 'CE'),
},
'N': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'OD1'),
},
'P': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'CD'),
},
'Q': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'CD'),
3: ('CB', 'CG', 'CD', 'OE1'),
},
'R': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'CD'),
3: ('CB', 'CG', 'CD', 'NE'),
4: ('CG', 'CD', 'NE', 'CZ'),
},
'S': {
1: ('N', 'CA', 'CB', 'OG'),
},
'T': {
1: ('N', 'CA', 'CB', 'OG1'),
},
'V': {
1: ('N', 'CA', 'CB', 'CG1'),
},
'W': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'CD1'),
},
'Y': {
1: ('N', 'CA', 'CB', 'CG'),
2: ('CA', 'CB', 'CG', 'CD1'),
},
}
Finally, it will be easier to look up the coordinates of atoms in the side chain by their indices rather than by their names. So, using the previous two mappings, we’ll create a mapping that links each amino acid to its chi angles and the corresponding atom indices that define these angles.
1
2
3
4
5
6
7
chi_idxs = {}
for aa in chi_atoms:
chi_idxs[aa] = {}
for i, atoms in chi_atoms[aa].items():
chi_idxs[aa][i] = [atom_order[aa].index(atom) for atom in atoms]
print(chi_idxs)
{'C': {1: [0, 1, 4, 5]},
'D': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6]},
'E': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6], 3: [4, 5, 6, 7]},
'F': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6]},
'H': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6]},
'I': {1: [0, 1, 4, 5], 2: [1, 4, 5, 7]},
'K': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6], 3: [4, 5, 6, 7], 4: [5, 6, 7, 8]},
'L': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6]},
'M': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6], 3: [4, 5, 6, 7]},
'N': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6]},
'P': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6]},
'Q': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6], 3: [4, 5, 6, 7]},
'R': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6], 3: [4, 5, 6, 7], 4: [5, 6, 7, 8]},
'S': {1: [0, 1, 4, 5]},
'T': {1: [0, 1, 4, 5]},
'V': {1: [0, 1, 4, 5]},
'W': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6]},
'Y': {1: [0, 1, 4, 5], 2: [1, 4, 5, 6]}}
Since chi angles are dihedral angles between planes formed by four atoms, we will define a helper function to calculate the dihedral angle between four points in 3D space. One convention for dihedral angles is to ensure the angle falls between -180 and 180 degrees. In this tutorial, however, we will use the convention of constraining the angle between 0 and 360 degrees. Using some trigonometry, we can calculate the dihedral angle between four points in 3D space as follows:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
def get_dihedral(A, B, C, D) -> float:
"""Calculate the dihedral angle between points A, B, C, and D in the range [0, 360] using arctan2."""
AB = B - A
BC = C - B
CD = D - C
N1 = np.cross(AB, BC)
N2 = np.cross(BC, CD)
N1 /= np.linalg.norm(N1)
N2 /= np.linalg.norm(N2)
x = np.dot(N1, N2)
y = np.dot(np.cross(N1, N2), BC / np.linalg.norm(BC))
angle = np.degrees(np.arctan2(y, x))
if angle < 0:
angle += 360
return angle
Now we can define a wrapper function to calculate all the chi angles for a given protein residue. The output will be a 4-dimensional tensor, with a value of 0.0 for chi angles that do not exist for that amino acid.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
def get_res_coords(res):
"""Get the one-letter code and coordinates of atoms in a residue."""
res_code = seq1(res.get_resname())
coords = [None] * 14 # 14 atoms in the longest amino acid
for atom in res:
if atom.get_name() in atom_order[res_code]:
coords[atom_order[res_code].index(atom.get_name())] = atom.get_coord()
return res_code, coords
def get_chi_angles(res_code: str, coords: list) -> torch.Tensor:
"""Calculate the chi angles for a residue given its one-letter code and coordinates."""
chi_angles = [0.0] * 4 # 4 chi angles for the longest amino acid
if res_code in chi_atoms:
for i, idxs in chi_idxs[res_code].items():
chi_coords = [coords[idx] for idx in idxs]
chi_angles[i-1] = get_dihedral(*chi_coords)
return torch.tensor(chi_angles)
To see the function in action, let’s calculate the chi angles for the residues in the 5HMK protein and display them for the first 20 residues.
1
2
3
4
5
6
7
8
9
10
11
parser = PDBParser()
structure = parser.get_structure("5hmk", "data/raw/5hmk.pdb")
chi_angles = []
for chain in structure.get_chains():
for res in chain:
res_code, coords = get_res_coords(res)
chi_angles.append(get_chi_angles(res_code, coords))
chi_angles = torch.stack(chi_angles)
print(chi_angles[:20])
tensor([[299.9426, 0.0000, 0.0000, 0.0000],
[290.7721, 167.9033, 0.0000, 0.0000],
[299.6244, 0.0000, 0.0000, 0.0000],
[188.9346, 183.0718, 286.1503, 166.9276],
[335.1982, 38.9081, 0.0000, 0.0000],
[294.0338, 181.2074, 173.5386, 163.2308],
[350.6445, 354.1471, 0.0000, 0.0000],
[289.6092, 179.8937, 0.0000, 0.0000],
[177.2721, 65.4467, 0.0000, 0.0000],
[195.3459, 48.8548, 0.0000, 0.0000],
[ 95.5402, 187.5152, 217.7537, 186.4376],
[195.8226, 127.5637, 0.0000, 0.0000],
[251.1091, 36.3955, 0.0000, 0.0000],
[283.4535, 232.8311, 182.9514, 80.0497],
[ 81.9222, 0.0000, 0.0000, 0.0000],
[280.7255, 0.0000, 0.0000, 0.0000],
[ 0.0000, 0.0000, 0.0000, 0.0000],
[ 0.0000, 0.0000, 0.0000, 0.0000],
[206.2470, 85.4079, 25.2864, 0.0000],
[ 40.8765, 196.3349, 189.7616, 141.9614]], dtype=torch.float64)
Another commom feature for geometry-aware models is the relative position of atoms in the side chain. Specifically, we’re interested in the vectors between the alpha carbon (CA) and the nitrogen (N) and carbon (C) atoms of the peptide bond. These vectors can be calculated simply as the difference between the coordinates of the respective atoms. We’ll define functions to calculate these vectors for a given protein residue, as well as a function to output the coordinates of the alpha carbon, which we will use later.
1
2
3
4
5
6
7
8
9
10
11
def get_c_alpha_pos(coords: list) -> torch.Tensor:
"""Get the position of the alpha carbon of a residue given its coordinates."""
return torch.tensor(coords[1])
def get_n_rel_pos(coords: list) -> torch.Tensor:
"""Get the relative position of the backbone nitrogen of a residue given its coordinates."""
return torch.tensor(coords[0] - coords[1])
def get_c_rel_pos(coords: list) -> torch.Tensor:
"""Get the relative position of the backbone carbon of a residue given its coordinates."""
return torch.tensor(coords[2] - coords[1])
Let’s again use the 5HMK protein as an example and calculate c_alpha_pos, n_rel_pos, and c_rel_pos for the first 5 residues:
1
2
3
4
5
6
7
for i, res in enumerate(chain.get_residues()):
if i >= 5: break
res_code, coords = get_res_coords(res)
c_alpha_pos = get_c_alpha_pos(coords)
n_rel_pos = get_n_rel_pos(coords)
c_rel_pos = get_c_rel_pos(coords)
print(f'res: {res_code}, c_alpha_pos: {c_alpha_pos}, n_rel_pos: {n_rel_pos}, c_rel_pos: {c_rel_pos}')
res: T, c_alpha_pos: tensor([10.4830, -8.7710, 32.2980]), n_rel_pos: tensor([-0.2720, -0.9290, -1.1080]), c_rel_pos: tensor([-0.8650, 1.2670, -0.0920])
res: L, c_alpha_pos: tensor([ 8.1170, -5.9820, 33.5160]), n_rel_pos: tensor([ 0.8360, -1.1830, -0.1910]), c_rel_pos: tensor([0.8070, 1.0520, 0.7880])
res: V, c_alpha_pos: tensor([ 9.8000, -2.6010, 34.2800]), n_rel_pos: tensor([-0.7560, -1.1300, -0.5490]), c_rel_pos: tensor([-0.8980, 1.2330, 0.1970])
res: R, c_alpha_pos: tensor([ 8.5160, 0.6640, 35.8800]), n_rel_pos: tensor([ 0.6900, -1.2440, -0.3700]), c_rel_pos: tensor([ 1.0150, 1.1350, -0.1700])
res: P, c_alpha_pos: tensor([10.4370, 3.6740, 34.4220]), n_rel_pos: tensor([-0.9540, -1.1070, 0.1710]), c_rel_pos: tensor([-0.1000, 1.0220, 1.1210])
Protein Dataset
Now we can bring all the components together to create a PyTorch Geometric dataset that includes the ESM3 embeddings, chi angles, and relative positions of atoms in the side chains. We’ll use the torch_geometric.data.Data class to store data for each protein residue. The dataset will contain the following attributes for a protein with (N) residues:
name: The PDB ID of the protein.x: The ESM3 embeddings for each residue ([N, 1536]) concatenated with one-hot encodings of the amino acid ([N, 20]) –> total size of [N, 1556].pos: Coordinates of the alpha carbon atom in each residue ([N, 3]).sidechain_feats: Concatenation of the following features ([N, 10] in total):chi_angles: The chi angles for each residue ([N, 4]) normalized to the range [0, 1] by dividing by 360.n_rel_pos: Position of the backbone nitrogen atom relative to the alpha carbon in each residue ([N, 3]).c_rel_pos: Position of the backbone carbon atom relative to the alpha carbon in each residue ([N, 3]).
Let’s define the ProteinDataset class, which takes as input the path to the ESM3 embeddings and to the root directory containing the .pdb files in the raw subdirectory.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
class ProteinDataset(InMemoryDataset):
'''Custom protein dataset with ESM3 embeddings, one-hot encoded amino acids, chi angles, and relative positions.'''
url = 'https://github.com/vladislach/protein-dataset/raw/main/proteins.zip'
def __init__(self, root, esm_embs_path, transform=None):
self.esm_embs = torch.load(esm_embs_path)
self.pdb_ids = list(self.esm_embs.keys())
super().__init__(root, transform)
self.load(self.processed_paths[0])
def raw_file_names(self):
return [f"{pdb_id}.pdb" for pdb_id in self.pdb_ids]
def processed_file_names(self):
return ['data.pt']
def download(self):
path = download_url(self.url, self.raw_dir)
extract_zip(path, self.raw_dir)
os.unlink(path)
def process(self):
data_list = []
parser = PDBParser()
for pdb_id, path in zip(self.pdb_ids, self.raw_paths):
structure = parser.get_structure(pdb_id, path)
seq = ''
chi_angles, c_alpha_pos, n_rel_pos, c_rel_pos = [], [], [], []
for chain in structure.get_chains():
for res in chain:
if not is_aa(res): continue
res_code, coords = self.get_res_coords(res)
chi_angles.append(self.get_chi_angles(res_code, coords) / 360)
c_alpha_pos.append(self.get_c_alpha_pos(coords))
n_rel_pos.append(self.get_n_rel_pos(coords))
c_rel_pos.append(self.get_c_rel_pos(coords))
seq += res_code
esm_emb = self.esm_embs[pdb_id]
res_one_hot = one_hot(torch.tensor([amino_acids.index(aa) for aa in seq]), len(amino_acids), dtype=torch.float)
chi_angles = torch.stack(chi_angles)
c_alpha_pos = torch.stack(c_alpha_pos)
n_rel_pos = torch.stack(n_rel_pos)
c_rel_pos = torch.stack(c_rel_pos)
data = Data(
name=pdb_id,
x=torch.cat([res_one_hot, esm_emb], dim=-1),
pos=c_alpha_pos,
sidechain_feats=torch.cat([chi_angles, n_rel_pos, c_rel_pos], dim=-1)
)
data_list.append(data)
self.save(data_list, self.processed_paths[0])
def get_res_coords(self, res):
"""Get the coordinates of the atoms of a given residue."""
res_code = seq1(res.get_resname())
coords = [None] * 14
for atom in res:
if atom.get_name() in atom_order[res_code]:
coords[atom_order[res_code].index(atom.get_name())] = atom.get_coord()
return res_code, coords
def get_dihedral(self, A, B, C, D) -> float:
"""Calculate the dihedral angle between points A, B, C, and D in the range [0, 360] using arctan2."""
AB = B - A
BC = C - B
CD = D - C
N1 = np.cross(AB, BC)
N2 = np.cross(BC, CD)
N1 /= np.linalg.norm(N1)
N2 /= np.linalg.norm(N2)
x = np.dot(N1, N2)
y = np.dot(np.cross(N1, N2), BC / np.linalg.norm(BC))
angle = np.degrees(np.arctan2(y, x))
if angle < 0:
angle += 360
return angle
def get_chi_angles(self, res_code: str, coords: list) -> torch.Tensor:
"""Calculate the chi angles for a residue given its one-letter code and coordinates."""
chi_angles = [0.0] * 4
if res_code in chi_atoms:
for i, idxs in chi_idxs[res_code].items():
chi_coords = [coords[idx] for idx in idxs]
chi_angles[i-1] = get_dihedral(*chi_coords)
return torch.tensor(chi_angles)
def get_c_alpha_pos(self, coords: list) -> torch.Tensor:
"""Get the position of the alpha carbon of a residue given its coordinates."""
return torch.tensor(coords[1])
def get_n_rel_pos(self, coords: list) -> torch.Tensor:
"""Get the relative position of the backbone nitrogen of a residue given its coordinates."""
return torch.tensor(coords[0] - coords[1])
def get_c_rel_pos(self, coords: list) -> torch.Tensor:
"""Get the relative position of the backbone carbon of a residue given its coordinates."""
return torch.tensor(coords[2] - coords[1])
Finally, let’s create an instance of the ProteinDataset class and take a look at the datapoints we have created:
1
2
3
4
dataset = ProteinDataset('data', 'data/esm_embs.pt')
for data in dataset:
print(data)
Data(x=[257, 1556], pos=[257, 3], name='4q87', sidechain_feats=[257, 10])
Data(x=[86, 1556], pos=[86, 3], name='5hmk', sidechain_feats=[86, 10])
Data(x=[163, 1556], pos=[163, 3], name='3dpf', sidechain_feats=[163, 10])
Data(x=[603, 1556], pos=[603, 3], name='6h77', sidechain_feats=[603, 10])
Data(x=[547, 1556], pos=[547, 3], name='3a2c', sidechain_feats=[547, 10])
Data(x=[279, 1556], pos=[279, 3], name='2zy1', sidechain_feats=[279, 10])
The attribute shapes are as expected, and we have successfully created a PyTorch Geometric dataset containing the ESM3 embeddings, chi angles, and relative positions of some atoms in the side chains of protein residues! This dataset is now ready for training geometry-aware models in protein structure prediction tasks.